Verordnung (EG) Nr. 648/2004 des Europäischen Parlaments und des Rates vom 31. März 2004 über Detergenzien
ANHANG VIII
Die Mitgliedstaaten wenden folgende Prüf- und Analysemethoden zur Kontrolle der im Verkehr befindlichen Detergenzien an:
1. Referenzmethode (Bestätigungstest)
1.1. Definition
Gegenstand dieser Methode ist ein Labormodell einer Belebtschlammanlage + Nachklärbecken als Simulation der kommunalen Abwasserbehandlung. Die beschriebenen Bedingungen stammen aus Richtlinien, die dieser Verordnung vorausgingen. Auf diese Prüfmethode können verbesserte Bedingungen nach dem neuesten Stand der Technik gemäß EN ISO 11733 angewendet werden.
1.2. Erforderliche Ausrüstung
Die Messung erfolgt unter Verwendung einer Belebtschlammanlage, die in Abbildung 1 schematisch und in Abbildung 2 ausführlicher dargestellt ist. Die Ausrüstung besteht aus einem Vorratsgefäß A für die synthetischen Abwässer, einer Dosierpumpe B, einem Belüftungsgefäß C, einem Absetzgefäß D, einer Druckluftpumpe E für den Belebtschlammrücklauf und einem Sammelgefäß F für das ablaufende behandelte Abwasser.
Die Gefäße A und F müssen aus Glas oder geeignetem Kunststoff bestehen und mindestens vierundzwanzig Liter fassen. Die Pumpe B muss einen gleichmäßigen Zufluss des synthetischen Abwassers zum Belüftungsgefäß gewährleisten; im normalen Betrieb enthält dieses Gefäß drei Liter Mischflüssigkeit. Im Gefäß C ist in der Spitze des konisch geformten Gefäßbodens eine Glasfilterfritte G zur Belüftung aufgehängt. Die Menge der durch die Fritte eingeblasenen Luft muss mit einem Mengenmessgerät H gemessen werden.
1.3. Synthetisches Abwasser
Zur Durchführung des Tests ist ein synthetisches Abwasser zu verwenden. Hierzu werden pro Liter Trinkwasser gelöst:
Das synthetische Abwasser wird täglich frisch hergestellt.
1.4. Herstellung der Proben
Unvermischte grenzflächenaktive Substanzen werden ohne Vorbehandlung getestet. Zur Herstellung des synthetischen Abwassers (Nummer 1.3) muss der aktive Gehalt der Tensidproben bestimmt werden.
1.5. Betrieb der Prüfeinrichtung
Zu Beginn des Tests werden das Belüftungsgefäß C sowie das Absetzgefäß D mit synthetischem Abwasser gefüllt. Das Absetzgefäß D wird in der Höhe so fixiert, dass das Belüftungsgefäß C drei Liter aufnimmt.
Die Impfung erfolgt mit 3 ml eines Kläranlagenablaufs guter Qualität, der frisch dem Ablauf einer biologischen Kläranlage für vorwiegend häusliches Abwasser entnommen wird. Die Ablaufprobe muss von der Entnahme bis zur Verwendung unter aeroben Bedingungen gehalten werden. Dann sind die Luftzufuhr G, die Druckluftpumpe E und die Dosierpumpe B einzuschalten. Der Zulauf des synthetischen Abwassers in das Belüftungsgefäß C muss einen Liter je Stunde betragen, was einer durchschnittlichen Aufenthaltszeit von drei Stunden entspricht.
Die Luftzufuhr ist so einzustellen, dass der Inhalt des Belüftungsgefäßes C ständig in Suspension verbleibt und ein Mindestgehalt an gelöstem Sauerstoff von 2 mg/l aufrechterhalten wird. Schaumbildung ist durch geeignete Maßnahmen zu verhindern. Jedoch dürfen keine Entschäumer verwendet werden, die eine hemmende Wirkung auf den Belebtschlamm ausüben oder Tenside enthalten. Die Pumpe E muss so eingestellt sein, dass stets ein gleichmäßiger Rücklauf von Belebtschlamm aus dem Absetzgefäß D zum Belüftungsgefäß C erfolgt. Der im oberen Teil des Belüftungsgefäßes C, am Boden des Absetzgefäßes D oder in der Rücklaufleitung sich ansammelnde Schlamm muss mindestens einmal täglich durch Bürsten oder durch andere geeignete Maßnahmen in den Umlauf zurückgebracht werden. Wenn der Schlamm sich nicht absetzt, kann sein Absetzverhalten durch gegebenenfalls wiederholte Zugabe von je 2 ml einer 5%igen Eisen(III)chloridlösung verbessert werden.
Das aus dem Absetzgefäß D abfließende Wasser wird in dem Sammelgefäß F während vierundzwanzig Stunden aufgefangen; nach Ablauf dieser Zeit wird nach gründlichem Durchmischen eine Probe entnommen. Anschließend ist das Sammelgefäß F sorgfältig zu reinigen.
1.6. Überwachung der Messanordnung
Der Tensidgehalt des synthetischen Abwassers (in mg/l) wird unmittelbar vor dem Gebrauch bestimmt.
Der Tensidgehalt (in mg/l) des im Sammelgefäß F während vierundzwanzig Stunden aufgefangenen Ablaufs wird analytisch nach derselben Methode unmittelbar nach der Probenahme bestimmt; andernfalls muss die Probe konserviert werden (vorzugsweise durch Einfrieren). Die Konzentration ist auf 0,1 mg/l Tensid genau zu bestimmen.
Zur Überwachung des einwandfreien Betriebs der Messanordnung wird mindestens zweimal wöchentlich der chemische Sauerstoffbedarf (CSB) oder der gelöste organische Kohlenstoff (DOC) des glasfasergefilterten Abwassers im Sammelgefäß F und des gefilterten synthetischen Abwassers im Vorratsgefäß A gemessen.
Nach Erreichen eines pro Tag nahezu gleich bleibenden biologischen Abbaus des Tensids, d. h. nach Ende der Einarbeitungszeit gemäß Abbildung 3, sollte die Verringerung des CSB oder DOC weitgehend stetig verlaufen.
Der Trockensubstanzgehalt des Belebtschlamms in g/l im Belüftungsgefäß ist zweimal wöchentlich zu ermitteln. Ist er größer als 2,5 g/l, so ist der Überschuss an Belebtschlamm zu entfernen.
Der Abbautest wird bei Raumtemperatur durchgeführt; diese sollte im Bereich zwischen 19 und 24 °C annähernd gleich bleiben.
1.7. Berechnung der biologischen Abbaubarkeit
Der biologische Abbau des Tensids in Prozenten ist täglich aus dem Tensidgehalt in mg/l des synthetischen Abwassers und des im entsprechenden Sammelgefäß F gesammelten Ablaufs zu errechnen.
Die so erhaltenen Abbaubarkeitswerte werden entsprechend Abbildung 3 grafisch dargestellt. Die Abbaubarkeit des Tensids ist als arithmetisches Mittel aus den Abbauwerten zu berechnen, die nach dem Ende der Einlauf- und Akklimatisierungszeit an einundzwanzig aufeinander folgenden Tagen bei gleich bleibendem Abbau in störungsfreiem Betrieb ermittelt wurden. In keinem Fall soll die Einlaufzeit länger als sechs Wochen dauern.
Die täglichen biologischen Abbauwerte werden bis auf 0,1 % genau berechnet; das Endergebnis ist jedoch auf ganze Zahlen auf- bzw. abzurunden.
In manchen Fällen kann die Häufigkeit der Probenahmen beschränkt werden, jedoch sind zur Ermittlung des Mittelwerts mindestens vierzehn Ergebnisse zu verwenden, die innerhalb von einundzwanzig Tagen nach der Einlaufzeit erhalten wurden.
2. Bestimmung anionischer Tenside in Prüfungen zur biologischen Abbaubarkeit
2.1. Grundsatz
Das Verfahren beruht auf der Tatsache, dass der kationische Farbstoff Methylenblau mit anionischen Tensiden (MBAS) blaue Salze bildet, die mit Chloroform extrahiert werden können. Zur Vermeidung von Störungen erfolgt die Extraktion zuerst aus alkalischer Lösung; sodann wird der Extrakt mit saurer Methylenblaulösung geschüttelt. Die Extinktion der abgetrennten organischen Phase wird fotometrisch im Absorptionsmaximum bei einer Wellenlänge von 650 nm gemessen.
2.2. Chemikalien und Geräte
2.2.1. Pufferlösung pH 10
24 g Natriumhydrogencarbonat (NaHCO3 ) p.a. und 27 g wasserfreies Natriumcarbonat (Na2CO3) p.a. in entionisiertem Wasser lösen und auf 1 000 ml verdünnen.
2.2.2. Neutrale Methylenblaulösung
0,35 g Methylenblau p.a. in entionisiertem Wasser lösen und auf 1 000 ml verdünnen. Diese Lösung mindestens vierundzwanzig Stunden vor Gebrauch zubereiten. Die Extinktion der Chloroformphase der Blindprobe darf gegenüber reinem Chloroform bei 650 nm 0,015 je cm Schichtdicke nicht übersteigen.
2.2.3. Saure Methylenblaulösung
0,35 g Methylenblau p.a. in 500 ml entionisiertem Wasser auflösen und mit 6,5 ml H2SO4 (D = 1,84 g/ml) mischen. Mit entionisiertem Wasser auf 1 000 ml verdünnen. Diese Lösung mindestens vierundzwanzig Stunden vor Gebrauch zubereiten. Die Extinktion der Chloroformphase der Blindprobe darf gegenüber reinem Chloroform bei 650 nm 0,015 je cm Schichtdicke nicht übersteigen.
| 2.2.4. | Chloroform (Trichlormethan) p.a. (frisch destilliert) |
| 2.2.5. | Dodecylbenzolsulfonsäuremethylester |
| 2.2.6. | Ethanolische Kaliumhydroxidlösung (KOH 0,1 M) |
| 2.2.7. | Ethanol, rein, C2H5OH |
| 2.2.8. | Schwefelsäure, H2SO4, 0,5 M |
| 2.2.9. | Phenolphthaleinlösung1 g Phenolphthalein in 50 ml Ethanol und 50 ml entionisiertem Wasser unter ständigem Umrühren lösen. Jedweden Niederschlag abfiltrieren. |
| 2.2.10. | Methanolische Salzsäure (250 ml Salzsäure p.a. und 750 ml Methanol) |
| 2.2.11. | Scheidetrichter, 250 ml |
| 2.2.12. | Messkolben, 50 ml |
| 2.2.13. | Messkolben, 500 ml |
| 2.2.14. | Messkolben, 1 000 ml |
| 2.2.15. | Rundkolben mit Schliff und Rückflusskühler, 250 ml; Siedeperlen |
| 2.2.16. | pH-Meter |
| 2.2.17. | Fotometer für Messungen bei 650 nm mit 1- bis 5-cm-Küvetten |
| 2.2.18. | Grobporiges Filterpapier |
2.3. Verfahren
Die Analyseproben dürfen nicht durch eine Schaumschicht entnommen werden.
Nach eingehender Reinigung mit Wasser sind die Analysegeräte vor Verwendung gründlich mit methanolischer Salzsäure (2.2.10) und anschließend mit entionisiertem Wasser zu spülen.
Proben des zu prüfenden Zu- und Abflusses der Belebtschlammanlage bei der Probeentnahme sofort filtrieren. Die ersten 100 ml des Filtrats werden verworfen.
Eine abgemessene Probemenge, gegebenenfalls nach Neutralisierung, in einen 250-ml-Scheidetrichter (2.2.11) geben. Die Probemenge sollte 20 bis 150 μg MBAS enthalten. Bei niedrigerem MBAS-Gehalt können bis zu 100 ml Probe benutzt werden. Werden weniger als 100 ml verwendet, so ist mit entionisiertem Wasser auf 100 ml zu verdünnen. 10 ml Pufferlösung (2.2.1), 5 ml neutrale Methylenblaulösung (2.2.2) und 15 ml Chloroform (2.2.4) zur Probe hinzufügen. Gemisch eine Minute gleichmäßig und nicht zu stark schütteln. Nach Phasentrennung Chloroformschicht in einen zweiten Trenntrichter geben, der 110 ml entionisiertes Wasser und 5 ml saure Methylenblaulösung (2.2.3) enthält. Gemisch eine Minute schütteln. Chloroformschicht durch einen vorher mit Chloroform gewaschenen und benetzten Wattefilter in einen Messkolben geben (2.2.12).
Alkalische und saure Lösungen dreimal extrahieren, wobei bei der zweiten und dritten Extraktion je 10 ml Chloroform zu verwenden sind. Kombinierte Chloroformextrakte durch denselben Wattefilter filtrieren und bis zur Marke des 50-ml-Kolbens (2.2.12) mit dem zum Nachwaschen der Watte benutzten Chloroform verdünnen. Extinktion der Chloroformlösung gegenüber Chloroform bei 650 nm in 1- bis 5-cm-Küvetten messen. Das ganze Verfahren mit einem Blindversuch durchführen.
2.4. Eichkurve
Aus der Standardsubstanz Dodecylbenzolsulfonsäuremethylester (Tetrapropylen-Typ, MG 340) nach Verseifung zum Kaliumsalz eine Eichlösung herstellen. Die MBAS wird als Natriumdodecylbenzolsulfonat (MG 348) berechnet.
Mit einer Messpipette 400 bis 450 mg Dodecylbenzolsulfonsäuremethylester (2.2.5) auf 0,1 mg genau in einen Rundkolben einwiegen und 50 ml ethanolische Kaliumhydroxidlösung (2.2.6) und einige Siedeperlen hinzugeben. Rückflusskühler anbringen und eine Stunde lang kochen. Nach Abkühlung Kühler und Schliff mit 30 ml Ethanol waschen und die Waschflüssigkeit zum Kolbeninhalt hinzugeben. Lösung mit Schwefelsäure gegenüber Phenolphthalein bis zur Farblosigkeit titrieren. Diese Lösung in einen 1 000 -ml-Messkolben (2.2.14) umgießen, bis zur Marke mit entionisiertem Wasser nachfüllen und mischen.
Ein Teil dieser Tensid-Stammlösung ist dann weiter zu verdünnen. 25 ml entnehmen, in einen 500-ml-Messkolben (2.2.13) geben, mit entionisiertem Wasser bis zur Marke nachfüllen und mischen.
Diese Standardlösung enthält:
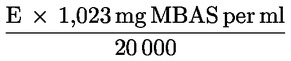
wobei E das Gewicht der Probe in mg bedeutet.
Zur Festlegung der Eichkurve sind je 1, 2, 4, 6 und 8 ml Standardlösung zu entnehmen und mit entionisiertem Wasser auf jeweils 100 ml zu verdünnen. Anschließend wie in Nummer 2.3 beschrieben (einschließlich des Blindtests) verfahren.
2.5. Berechnung der Ergebnisse
Der Gehalt an anionischen Tensiden (MBAS) in der Probe wird aus der Eichkurve (2.4) abgelesen. Der MBASGehaltder Probe ergibt sich aus:
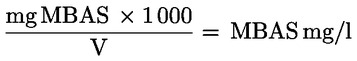
wobei V das Volumen der benutzten Probe in ml bedeutet.
Die Ergebnisse sind als Natriumdodecylbenzolsulfonat (MG 348) anzugeben.
2.6. Angabe der Ergebnisse
Die Ergebnisse sind in mg/l MBAS auf 0,1 genau anzugeben.
3. Prüfungen zur biologischen Abbaubarkeit: Bestimmung nichtionischer Tenside in Prüfflüssigkeiten
3.1. Grundsatz
Grenzflächenaktive Substanzen werden konzentriert und durch Ausblasen abgetrennt. In der eingesetzten Probemenge sollte der Gehalt an nichtionischen Tensiden im Bereich zwischen 250 und 800 μg liegen.
Das ausgeblasene Tensid wird in Ethylacetat gelöst.
Nach Phasentrennung und Eindampfen des Lösungsmittels wird das nichtionische Tensid in wässriger Lösung mit modifiziertem Dragendorffschen Reagens (KBiI4 + BaCl2 + Eisessig) gefällt.
Der Niederschlag wird abfiltriert, mit Eisessig gewaschen und in Ammoniumtartratlösung gelöst. Das in Lösung befindliche Bismut wird bei pH 4-5 mit Pyrrolidindithiocarbamatlösung unter Verwendung einer blanken Platinindikatorelektrode und einer Kalomel- oder Silber/Silberchlorid-Referenzelektrode potenziometrisch titriert. Die Methode ist auf nichtionische Tenside mit 6-30 Alkylenoxidgruppen anwendbar.
Das Titrationsergebnis wird mit dem empirischen Eichfaktor 54 zur Umrechnung auf die Bezugssubstanz Nonylphenol mit 10 Molen Ethylenoxid (NP 10) multipliziert.
3.2. Chemikalien und Geräte
Alle wässrigen Lösungen sind mit entionisiertem Wasser herzustellen.
| 3.2.1. | Reines Ethylacetat, frisch destilliert |
| 3.2.2. | Natriumhydrogencarbonat (NaHCO3 ) p.a. |
| 3.2.3. | Verdünnte Salzsäure (HCl) [20 ml konzentrierte Salzsäure p.a., mit Wasser auf 1 000 ml auffüllen] |
| 3.2.4. | Methanol p.a., frisch destilliert, in Glasflaschen aufbewahrt |
| 3.2.5. | Bromkresolpurpur, 0,1 g in 100 ml Methanol |
| 3.2.6. | Fällungsreagenz: Das Fällungsreagenz ist eine Mischung von 2 Volumenteilen der Lösung A und 1 Volumenteil der Lösung B. Die Mischung ist in einer braunen Flasche aufzubewahren und bis zu einer Woche haltbar.3.2.6.1. Lösung A 1,7 g Bismut(III)nitrat p.a. (BiONO3.H2O) werden in 20 ml Eisessig gelöst und mit Wasser auf 100 ml aufgefüllt. Dann werden 65 g Kaliumiodid p.a. in 200 ml Wasser gelöst. Diese beiden Lösungen werden in einem 1 000 -ml-Messkolben gemischt, 200 ml Eisessig (3.2.7) hinzugefügt und mit Wasser bis zur Marke aufgefüllt. 3.2.6.2. Lösung B 290 g Bariumchlorid (BaCl2.2H2O) p.a. werden in 1 000 ml Wasser gelöst. |
| 3.2.7. | Eisessig, 99-100%ig (Essigsäure geringerer Konzentration ist ungeeignet). |
| 3.2.8. | Ammoniumtartratlösung: 12,4 g Weinsäure p.a. und 12,4 ml Ammoniaklösung p.a. (D = 0,910 g/ml) werden gemischt und mit Wasser auf 1 000 ml aufgefüllt (oder eine gleiche Menge von Ammoniumtartrat p.a. verwenden). |
| 3.2.9. | Ammoniaklösung: 40 ml Ammoniaklösung p.a. (D = 0,910 g/ml) werden mit Wasser auf 1 000 ml aufgefüllt. |
| 3.2.10. | Standardacetatpufferlösung: 40 g Natriumhydroxid p.a. werden in ein Becherglas mit etwa 500 ml Wasser gegeben, gelöst und abgekühlt. Dann werden 120 ml Eisessig (3.2.7) zugefügt. Nach gründlichem Mischen und Abkühlen wird die Lösung in einen 1 000 -ml-Messkolben umgefüllt. Man füllt mit Wasser bis zur Marke auf. |
| 3.2.11. | Pyrrolidindithiocarbamatlösung (nachstehend „Carbatlösung“ genannt): Man löst 103 mg Pyrrolidindithiocarbonsäure-Natriumsalz (C5H8NNaS2.2H2O) in etwa 500 ml Wasser, gibt 10 ml n-Amylalkohol p.a. und 0,5 g Natriumhydrogencarbonat p.a. (NaHCO3) hinzu und füllt mit Wasser auf 1 000 ml auf. |
| 3.2.12. | Kupfersulfatlösung (für die Eichung der Lösung 3.2.11).STAMMLÖSUNG 1,249 g Kupfersulfat p.a. (CuSO4.5H2O) werden mit 50 ml 0,5 M Schwefelsäure gemischt und zu 1 000 ml mit Wasser aufgefüllt. STANDARDLÖSUNG 50 ml der Stammlösung und 10 ml 0,5 M H2SO4 werden gemischt und mit Wasser zu 1 000 ml aufgefüllt. |
| 3.2.13. | Natriumchlorid p.a. |
| 3.2.14. | Tensid-Ausblasegerät (siehe Abbildung 5)Der Durchmesser der Glasfilterfritte und der Innendurchmesser des Zylinders müssen gleich groß sein. |
| 3.2.15. | Trenntrichter, 250 ml |
| 3.2.16. | Magnetrührwerk mit Magnetstab 25-30 mm |
| 3.2.17. | Goochtiegel, Durchmesser des perforierten Bodens 25 mm, Typ G4 |
| 3.2.18. | Rundfilter aus Glasfaserpapier, Durchmesser 27 mm, Faserdurchmesser 0,3-1,5 μm. |
| 3.2.19. | Zwei Saugflaschen mit Anschlussstutzen und Gummimanschette, Inhalt je 250 und 500 ml. |
| 3.2.20. | Registrierendes Potenziometer mit einer blanken Platinindikatorelektrode und einer Kalomel- oder Silber/Silberchlorid-Referenzelektrode, Messbereich 250 mV, mit automatischer Bürette von 20-25 ml Inhalt oder alternativ eine entsprechende manuelle Einrichtung. |
3.3. Methode
3.3.1. Anreicherung und Isolierung der grenzflächenakiven Substanzen
Die wässrige Probe wird durch ein grobporiges Filter filtriert. Die ersten 100 ml des Filtrats werden verworfen.
In das zuvor mit Ethylacetat durchgespülte Ausblasegerät wird eine abgemessene Probemenge gegeben, die zu 250 bis 800 μg nichtionische Tenside enthalten soll.
Zur Verbesserung des Trenneffekts werden 100 g Natriumchlorid und 5 g Natriumhydrogencarbonat hinzugegeben.
Überschreitet das Probevolumen 500 ml, so werden diese Salze in fester Form in das Ausblasegerät gegeben und unter Durchleiten von Stickstoff oder Luft gelöst.
Kommt ein geringeres Probevolumen zur Anwendung, werden diese Salze in etwa 400 ml Wasser gelöst und dann zugegeben.
Mit Wasser bis zum oberen Ablasshahn auffüllen.
Über die wässrige Phase werden vorsichtig 100 ml Ethylacetat aufgegeben.
Die Waschflasche in der Gasstromzuleitung (Stickstoff oder Luft) wird zu etwa zwei Drittel mit Ethylacetat gefüllt.
Man leitet einen Gasstrom von 30 bis 60 l je Stunde durch die Apparatur; die Verwendung eines Strömungsmessers ist zu empfehlen. Der Gasdurchsatz wird anfangs schrittweise erhöht. Die Gasmenge muss so bemessen sein, dass die Phasen erkennbar getrennt bleiben und eine Vermischung der Phasen und ein Auflösen des Ethylacetats im Wasser möglichst vermieden wird. Nach fünf Minuten wird der Gasstrom abgestellt.
Ist das Volumen der organischen Phase durch Lösen in Wasser um mehr als 20 % vermindert worden, so ist das Ausblasen unter Verringerung des Gasdurchsatzes zu wiederholen.
Die organische Phase wird in einen Scheidetrichter abgelassen. Die im Scheidetrichter gegebenenfalls abgesetzte wässrige Phase — es sollten nur wenige ml sein — wird in das Ausblasegerät zurückgegeben. Die Ethylacetat-Phase wird durch ein trockenes, grobporiges Filterpapier in ein 250-ml-Becherglas filtriert.
Man gibt erneut 100 ml Ethylacetat in das Ausblasegerät und leitet weitere fünf Minuten lang Stickstoff oder Luft hindurch. Die organische Phase wird in den bereits bei der ersten Abtrennung benutzten Scheidetrichter abgelassen. Die wässrige Phase wird verworfen und die organische Phase über das gleiche Filter wie die erste Ethylacetatmenge gegeben. Scheidetrichter und Filter werden mit 20 ml Ethylacetat nachgespült.
Der Ethylacetat-Extrakt wird unter Verwendung eines Wasserbads unter dem Abzug bis zur Trockne eingedampft. Zur Beschleunigung der Verdunstung wird auf die Oberfläche der Lösung ein leichter Luftstrom gerichtet.
3.3.2. Fällen und Filtrieren
Der nach 3.3.1 erhaltene Trockenrückstand wird in 5 ml Methanol gelöst, dann werden 40 ml Wasser und 0,5 ml verdünnte Salzsäure (3.2.3) hinzugegeben und die Lösung mit einem Magnetrührer durchgerührt.
In diese Lösung gibt man aus einem Messzylinder 30 ml Fällungsreagenz (3.2.6) hinzu. Der Niederschlag bildet sich bei fortgesetztem Rühren. Nach zehnminütigem Rühren wird die Mischung für mindestens fünf Minuten stehen gelassen.
Danach filtriert man die Mischung durch einen Gooch-Tiegel, dessen Boden mit einem Glasfaser-Filterpapier belegt ist. Das Filter wird zuvor mit etwa 2 ml Eisessig angefeuchtet und dabei angesaugt. Becherglas, Magnetstab und Tiegel werden gründlich mit Eisessig nachgewaschen, wozu etwa 40 bis 50 ml notwendig sind. Es ist nicht erforderlich, den am Becherglas fest anhaftenden Niederschlag quantitativ auf das Filter zu bringen, da die Lösung des Niederschlags vor der Filtration wieder in das Fällungs-Becherglas gegeben und der verbleibende Niederschlag dann gelöst wird.
3.3.3. Lösen des Niederschlags
Der Niederschlag im Filtertiegel wird durch Zugabe von heißer Ammoniumtartratlösung (etwa 80 °C) (3.2.8) in drei Portionen von je 10 ml gelöst. Jede Portion wird einige Minuten im Filtertiegel stehen gelassen, bevor sie durch das Filter in die Flasche abgesaugt wird.
Der Inhalt der Saugflasche wird in das Fällungs-Becherglas gegeben. Die Wand des Becherglases wird mit weiteren 20 ml Ammoniumtartratlösung gespült, um den Rest des Niederschlags zu lösen.
Filtertiegel, Anschlussstutzen und Saugflasche werden gründlich mit 150 bis 200 ml Wasser gewaschen und dieses Wasser in das Fällungs-Becherglas gegeben.
3.3.4. Titration
Man rührt die Lösung mit dem Magnetrührwerk (3.2.16), setzt einige Tropfen Bromkresolpurpurlösung (3.2.5) zu und stellt mit der verdünnten Ammoniaklösung (3.2.9) auf Farbumschlag nach violett ein (die Lösung ist durch Essigsäurereste, die vom Nachwaschen herrühren, zu Beginn schwach sauer).
Dann gibt man 10 ml Standardacetatpufferlösung (3.2.10) hinzu, führt die Elektroden in die Lösung ein und titriert mit eingetauchter Bürettenspitze potenziometrisch mit der Carbatlösung (3.2.11).
Die Titrationsgeschwindigkeit soll 2 ml/min nicht überschreiten.
Als Endpunkt gilt der Schnittpunkt der Tangenten, die man an die beiden Äste der Potenzialkurve legt.
Eine gelegentlich zu beobachtende Abflachung der Beugung der Potenzialkurve lässt sich durch sorgfältiges Reinigen der Platin-Elektrode (durch Schleifen mit Schmirgelpapier) beheben.
3.3.5. Blindversuch
Parallel zu den eigentlichen Bestimmungen läuft ein Blindversuch mit, bei dem 5 ml Methanol und 40 ml Wasser eingesetzt und nach Nummer 3.3.2 weiterverarbeitet werden. Der Verbrauch im Blindversuch sollte unter 1 ml Messlösung liegen, andernfalls bestehen Zweifel über die Reinheit der Reagenzien (Nummern 3.2.3, 3.2.7, 3.2.8, 3.2.9, 3.2.10), insbesondere durch ihren Gehalt an Schwermetallen; in diesem Fall sind die Reagenzien zu ersetzen. Das Ergebnis des Blindversuchs ist bei der Berechnung zu berücksichtigen.
3.3.6. Kontrolle des Faktors der Carbatlösung
Der Faktor der Carbatlösung wird bei Verwendung täglich bestimmt. Hierzu werden 10 ml der Kupfersulfat-Eichlösung (3.2.12) mit Carbatlösung nach Zugabe von 100 ml Wasser und 10 ml Standardacetatpuffer (3.2.10) titriert. Beträgt die verbrauchte Menge „a“ ml, so errechnet sich der Faktor „f“ wie folgt:
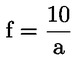
mit diesem Faktor sind die Titrationsergebnisse zu multiplizieren.
3.4. Berechnung der Ergebnisse
Jedes nichtionische Tensid hat einen von seiner Zusammensetzung, insbesondere von der Länge seiner Alkenoxidkette abhängigen Eichfaktor. Die Konzentration an nichtionischem Tensid wird im Verhältnis zu einer Referenzsubstanz ausgedrückt: diese ist ein Nonylphenol mit zehn Ethylenoxid-Einheiten (NP 10); der Umrechnungsfaktor hierfür ist gleich 0,054.
Die Menge des in der Probe vorhandenen Tensids lässt sich mit Hilfe dieses Faktors wie folgt berechnen (ausgedrückt in mg des NP-10-Äquivalents):
(b - c) × f × 0,054 = mg nichtionische Tenside als NP 10;
dabei ist
| b | = | der Verbrauch an Carbatlösung der Probe in ml, |
| c | = | der Verbrauch an Carbatlösung des Blindversuchs in ml, |
| f | = | der Faktor der Carbatlösung. |
3.5. Angabe der Ergebnisse
Die Ergebnisse sind in mg/l als NP 10 auf 0,1 genau anzugeben.
4. Vorbehandlung der zu prüfenden anionischen Tenside
4.1. Vorbemerkungen
4.1.1. Behandlung der Proben
Die Behandlung von anionischen grenzflächenaktiven Stoffen und von formulierten Detergenzien vor der Bestimmung der primären Bioabbaubarkeit durch den Bestätigungstest ist wie folgt:
| Erzeugnisse | Behandlung |
| Anionische Tenside | keine |
| Formulierte Detergenzien | alkoholische Extraktion, danach Isolierung der anionischen Tenside durch Ionenaustausch |
Zweck der alkoholischen Extraktion ist die Entfernung unlöslicher und anorganischer Bestandteile des kommerziellen Produkts, die den Test der biologischen Abbaubarkeit stören könnten.
4.1.2. Verfahren des Ionenaustauschs
Zur korrekten Durchführung des Tests der biologischen Abbaubarkeit ist die Isolierung und Abtrennung der anionischen Tenside von Seife, nichtionischen und kationischen Tensiden erforderlich.
Dies wird durch ein Ionenaustauschverfahren mittels eines makroporösen Anionenaustauscherharzes und geeigneter Elutionsmittel für fraktionierte Elution erreicht. Auf diese Weise werden Seife, anionische und nichtionische Tenside in einem einzigen Arbeitsgang isoliert.
4.1.3. Analytische Kontrolle
Der Gehalt an anionischen Tensiden in dem Wasch- und Reinigungsmittel wird nach Homogenisieren nach dem MBAS-Analysenverfahren bestimmt. Der Seifengehalt wird mittels einer geeigneten Analysenmethode bestimmt.
Diese Analyse des Produkts ist zur Berechnung der Mengen erforderlich, die zur Herstellung der Fraktionen für den Test der biologischen Abbaubarkeit erforderlich sind.
Eine quantitative Extraktion ist nicht erforderlich; doch sollten mindestens 80 % der anionischen Tenside extrahiert werden. In der Regel werden 90 % und mehr erhalten.
4.2. Grundsatz
Aus der homogenen Probe (Pulver, Pasten und vorher getrocknete Flüssigkeiten) wird ein Ethanolextrakt gewonnen, der die Tenside, die Seife und andere alkohollösliche Bestandteile der Wasch- und Reinigungsmittel-Probe enthält.
Der Ethanolextrakt wird bis zur Trockne verdampft, in Isopropanol-Wasser-Gemisch gelöst und diese Lösung durch eine auf 50 °C erhitzte Austauscherkombination aus stark saurem Kationenaustauscher und makroporösem Anionenaustauscher gegeben. Diese Temperatur ist erforderlich, um die Fällung von Fettsäuren im sauren Medium zu verhindern.
Die nichtionischen Tenside verbleiben im Filtrat.
Die Seifen-Fettsäuren werden durch Elution mit CO2-haltigem Ethanol abgetrennt. Die anionischen Tenside werden sodann durch Elution mit einer wässrigen Ammoniumhydrogencarbonat-Isopropanollösung als Ammoniumsalze erhalten. Diese Ammoniumsalze werden für den Abbaubarkeitstest verwendet.
Kationische Tenside, die den Abbaubarkeitstest und das Analysenverfahren stören könnten, werden durch den über dem Anionenaustauscher eingesetzten Kationenaustauscher entfernt.
4.3. Chemikalien und Geräte
4.4. Herstellung des Extrakts und Abtrennung der anionischen Tenside
4.4.1. Herstellung des Extrakts
Für den Abbaubarkeitstest ist eine Tensidmenge von etwa 50 g MBAS erforderlich.
Normalerweise werden nicht mehr als 1 000 g Produkt zur Extraktion eingesetzt, doch kann die Extraktion größerer Probemengen notwendig sein. Aus praktischen Gründen liegt die Höchstgrenze bei der Herstellung der Extrakte für den Abbaubarkeitstest in den meisten Fällen bei 5 000 g.
Erfahrungsgemäß ist die chargenweise Gewinnung der Extrakte arbeitstechnisch vorteilhafter als eine einmalige Extraktion einer größeren Menge. Die vorgeschriebenen Austauschermengen entsprechen einer Arbeitskapazität von 600 bis 700 mMol Tensiden und Seife.
4.4.2. Abtrennung der alkohollöslichen Bestandteile
250 g des zu untersuchenden Detergens werden in 1 250 ml Ethanol gegeben, das Gemisch wird eine Stunde unter Rühren und Rückfluss zum Sieden erhitzt. Die heiße alkoholische Lösung wird über eine auf 50 °C aufgeheizte Nutsche mit einem grobporigen Filter gegeben und rasch abfiltriert. Anschließend spült man Kolben und Nutsche mit rund 200 ml heißem Ethanol nach. Filtrat und Spülalkohol werden in einer Saugflasche aufgefangen.
Bei pastösen und flüssigen Produkten wägt man so viel ein, dass nicht mehr als 55 g anionisches Tensid und 35 g Seife vorliegen. Diese Einwaage wird bis zur Trockne gebracht. Der Rückstand wird in 2 000 ml Ethanol gelöst; dann wird wie vorstehend beschrieben verfahren.
Bei pulverförmigen Detergenzien mit geringerer Schüttdichte (< 300 g/l), ist es empfehlenswert, den Ethanolanteil auf ein Verhältnis von 20:1 zu erhöhen. Das ethanolische Filtrat wird — vorzugsweise mittels eines Rotationsverdampfers — zur Trockne eingedampft. Wird eine größere Extraktmenge benötigt, so wird das Verfahren wiederholt. Der Rückstand wird in 5 000 ml Isopropanol-Wasser-Gemisch gelöst.
4.4.3. Vorbereitung der Ionenaustauschersäulen
KATIONENAUSTAUSCHERSÄULE
600 ml KAT (4.3.6) werden in ein 3 000 -ml-Becherglas gegeben und mit 2 000 ml Salzsäure (4.3.8) übergossen.
Man lässt mindestens zwei Stunden unter gelegentlichem Umrühren stehen, sodann dekantiert man die Säure und spült den KAT mit entionisiertem Wasser in die Säule (4.3.12) ein, in die man zuvor einen Glaswollebausch eingelegt hat.
Die Säule wird mit entionisiertem Wasser bei einer Durchlaufgeschwindigkeit von 10-30 ml/min gewaschen, bis das Eluat chloridfrei ist.
Anschließend verdrängt man das Wasser mit 2 000 ml Isopropanol-Wasser-Gemisch (4.3.3), ebenfalls bei einer Durchlaufgeschwindigkeit von 10-30 ml/min. Damit ist die KAT-Säule betriebsbereit.
ANIONENAUSTAUSCHERSÄULE
600 ml AAT (4.3.7) werden in ein 3 000 -ml-Becherglas gegeben und mit 2 000 ml entionisiertem Wasser vollständig übergossen.
Dann lässt man den Austauscher mindestens zwei Stunden lang quellen.
Daraufhin spült man den KAT mit entionisiertem Wasser in die Säule ein. Die Säule sollte einen Glaswollebausch enthalten.
Die Säule wird mit 0,3 M Ammoniumhydrogencarbonatlösung (4.3.5) bis zur Chloridfreiheit gewaschen. Hierzu werden etwa 5 000 ml Lösung benötigt. Dann wird noch einmal mit 2 000 ml entionisiertem Wasser gewaschen. Anschließend verdrängt man das Wasser mit 2 000 ml Isopropanol-Wasser-Gemisch (4.3.3) bei einer Durchlaufgeschwindigkeit von 10-30 ml/min. Die AAT-Säule befindet sich nun in der OH-Form und ist betriebsbereit.
4.4.4. Verfahren des Ionenaustauschs
Man verbindet beide Austauschersäulen derart miteinander, dass sich die KAT-Säule oberhalb der AAT-Säule befindet.
Unter Verwendung eines Thermostaten werden die Austauschersäulen auf 50 °C aufgeheizt.
Dann werden 5 000 ml der nach Nummer 4.4.2 erhaltenen Lösung auf 60 °C erwärmt und die heiße Lösung mit einer Durchlaufgeschwindigkeit von 20 ml/min durch die Säulenkombination gegeben. Anschließend wäscht man mit 1 000 ml des heißen Isopropanol-Wasser-Gemisches (4.3.3) die Säulen nach.
Zur Gewinnung der anionischen Tenside (MBAS) wird die Kationensäule abgetrennt. Mit 5 000 ml Ethanol/CO2-Lösung bei 50 °C (4.3.4) Seifenfettsäuren aus der KAT-Säule eluieren. Eluat verwerfen.
Anschließend wird die MBAS mit 5 000 ml Ammoniumhydrogencarbonatlösung (4.3.5) aus der AAT-Säule eluiert und das Eluat im Dampfbad oder Rotationsverdampfer bis zur Trockne eingedampft.
Der Rückstand enthält die MBAS (als Ammoniumsalz) und möglicherweise nichttensidische anionische Stoffe, die den Test der biologischen Abbaubarkeit nicht beeinträchtigen. Bis zu einem bestimmten Volumen entionisiertes Wasser zum Rückstand hinzufügen und den MBAS-Gehalt in einem Aliquot bestimmen. Die Lösung wird als Standardlösung des anionischen Tensids für den Test der biologischen Abbaubarkeit verwendet. Sie ist bei einer Temperatur unter 5 °C aufzubewahren.
4.4.5. Regenerierung der verwendeten Austauscher
Der Kationenaustauscher wird nach Gebrauch verworfen.
Durch weitere Zugabe von Ammoniumhydrogencarbonatlösung (4.3.5) durch die Säule bei einer Durchflussgeschwindigkeit von etwa 10 ml/min, bis das Eluat von anionischen Tensiden frei ist (Methylenblau-Test), wird der Anionenaustauscher regeneriert.
Anschließend werden noch 2 000 ml Isopropanol-Wasser-Gemisch (4.3.3) durch den Anionenaustauscher gegeben. Danach ist der Anionenaustauscher wieder einsatzbereit.
5. Vorbehandlung der zu prüfenden nichtionischen Tenside
5.1. Vorbemerkungen
5.1.1. Behandlung der Proben
Die Behandlung von nichtionischen grenzflächenaktiven Stoffen und von formulierten Detergenzien vor der Bestimmung der biologischen Primärabbaubarkeit durch den Bestätigungstest ist wie folgt:
| Erzeugnisse | Behandlung |
| Nichtionische Tenside | keine |
| Formulierte Detergenzien | alkoholische Extraktion, danach Isolierung der nichtionischen Tenside durch Ionenaustausch |
Zweck der alkoholischen Extraktion ist die Entfernung unlöslicher und anorganischer Bestandteile des kommerziellen Produkts, die den Test der biologischen Abbaubarkeit stören könnten.
5.1.2. Verfahren des Ionenaustauschs
Zur korrekten Durchführung des Tests der biologischen Abbaubarkeit ist die Isolierung und Abtrennung der nichtionischen Tenside von Seife, anionischen und kationischen Tensiden erforderlich.
Dies wird durch ein Ionenaustauschverfahren mittels eines makroporösen Ionenaustauscherharzes und geeigneter Elutionsmittel für fraktionierte Elution erreicht. Auf diese Weise werden Seife, anionische und nichtionische Tenside in einem einzigen Arbeitsgang isoliert.
5.1.3. Analytische Kontrolle
Die Konzentration an anionischen und nichtionischen Tensiden in dem Wasch- und Reinigungsmittel wird nach Homogenisieren nach den MBAS- und BiAS-Analysenverfahren bestimmt. Der Seifengehalt wird mittels einer geeigneten Analysenmethode bestimmt.
Diese Analyse des Produkts ist zur Berechnung der Mengen erforderlich, die zur Herstellung der Fraktionen für den Test der biologischen Abbaubarkeit erforderlich sind.
Eine quantitative Extraktion ist nicht erforderlich; doch sollten mindestens 80 % der nichtionischen Tenside extrahiert werden. In der Regel werden 90 % und mehr erhalten.
5.2. Grundsatz
Aus der homogenen Probe (Pulver, Paste und vorher getrocknete Flüssigkeiten) wird ein Ethanolextrakt gewonnen, der die Tenside, die Seife und andere alkohollösliche Bestandteile der Wasch- und Reinigungsmittel-Probe enthält.
Der Ethanolextrakt wird bis zur Trockne verdampft, in Isopropanol-Wasser-Gemisch gelöst und diese Lösung durch eine auf 50 °C erhitzte Austauscherkombination aus stark saurem Kationenaustauscher und makroporösem Anionenaustauscher gegeben. Diese Temperatur ist erforderlich, um die Fällung von Fettsäuren im sauren Medium zu verhindern. Nach Eindampfen des Ablaufs erhält man die nichtionischen Tenside.
Kationische Tenside, die den Abbaubarkeitstest und das Analysenverfahren stören könnten, werden durch den über dem Anionenaustauscher eingesetzten Kationenaustauscher entfernt.
5.3. Chemikalien und Geräte
5.4. Herstellung des Extrakts und Abtrennung der nichtionischen Tenside
5.4.1. Herstellung des Extrakts
Für den Abbaubarkeitstest ist eine Tensidmenge von etwa 25 g MBAS erforderlich.
Bei der Herstellung der Extrakte für die Abbaubarkeitstests soll die einzusetzende Produktmenge auf höchstens 2 000 g beschränkt bleiben. Es kann daher nötig werden, die Aufarbeitung zweimal oder öfter durchzuführen, um die für den Abbaubarkeitstest genügende Menge zu erhalten.
Erfahrungsgemäß ist die chargenweise Gewinnung der Extrakte arbeitstechnisch vorteilhafter als eine einmalige Extraktion einer größeren Menge.
5.4.2. Abtrennung der alkohollöslichen Bestandteile
250 g des zu untersuchenden Detergens werden in 1 250 ml Ethanol gegeben, das Gemisch wird eine Stunde unter Rühren und Rückfluss zum Sieden erhitzt. Die heiße alkoholische Lösung wird über eine auf 50 °C aufgeheizte Nutsche mit einem grobporigen Filter gegeben und rasch abfiltiert. Anschließend spült man Kolben und Nutsche mit rund 200 ml heißem Ethanol nach. Filtrat und Spülalkohol werden in einer Saugflasche aufgefangen.
Bei pastösen und flüssigen Produkten wägt man so viel ein, dass nicht mehr als 25 g anionisches Tensid und 35 g Seife vorliegen. Diese Einwaage wird bis zur Trockne gebracht. Der Rückstand wird in 500 ml Ethanol gelöst; dann wird wie vorstehend beschrieben verfahren.
Bei pulverförmigen Detergenzien mit geringerer Schüttdichte (< 300 g/l), ist es empfehlenswert, den Ethanolanteil auf ein Verhältnis von 20:1 zu erhöhen. Das ethanolische Filtrat wird — vorzugsweise mittels eines Rotationsverdampfers — bis zur Trockne eingedampft. Wird eine größere Extraktmenge benötigt, so wird das Verfahren wiederholt. Der Rückstand wird in 5 000 ml Isopropanol-Wasser-Gemisch gelöst.
5.4.3. Vorbereitung der Ionenaustauschersäulen
KATIONENAUSTAUSCHERSÄULE
600 ml KAT (5.3.5) werden in ein 3 000 ml-Becherglas gegeben und mit 2 000 ml Salzsäure (5.3.7) übergossen. Man lässt mindestens zwei Stunden unter gelegentlichem Umrühren stehen, sodann dekantiert man die Säure und spült den KAT mit entionisiertem Wasser in die Säule (5.3.11) ein.
Die Säule sollte einen Glaswollebausch enthalten. Die Säule wird mit entionisiertem Wasser bei einer Durchlaufgeschwindigkeit von 10-30 ml/min gewaschen, bis das Eluat chloridfrei ist.
Anschließend verdrängt man das Wasser mit 2 000 ml Isopropanol-Wasser-Gemisch (5.3.3), ebenfalls bei einer Durchlaufgeschwindigkeit von 10-30 ml/min. Damit ist die KAT-Säule betriebsbereit.
ANIONENAUSTAUSCHERSÄULE
600 ml AAT (5.3.6) werden in ein Becherglas gegeben und mit 2 000 ml entionisiertem Wasser vollständig übergossen. Dann lässt man den Austauscher mindestens zwei Stunden lang quellen. Daraufhin spült man den KAT mit entionisiertem Wasser in die Säule ein. Die Säule sollte einen Glaswollebausch enthalten.
Die Säule wird mit 0,3 M Ammoniumhydrogencarbonatlösung (5.3.4) bis zur Chloridfreiheit gewaschen. Hierzu werden etwa 5 000 ml Lösung benötigt. Dann wird noch einmal mit 2 000 ml entionisiertem Wasser gewaschen.
Anschließend verdrängt man das Wasser mit 2 000 ml Isopropanol-Wasser-Gemisch (5.3.3), bei einer Durchlaufgeschwindigkeit von 10-30 ml/min. Die AAT-Säule befindet sich nun in der OH-Form und ist betriebsbereit.
5.4.4. Verfahren des Ionenaustauschs
Man verbindet beide Austauschersäulen derart miteinander, dass sich die KAT-Säule oberhalb der AAT-Säule befindet. Unter Verwendung eines Thermostaten werden die Austauschersäulen auf 50 °C aufgeheizt. Dann werden 5 000 ml der nach Nummer 5.4.2 erhaltenen Lösung auf 60 °C erwärmt und die heiße Lösung mit einer Durchlaufgeschwindigkeit von 20 ml/min durch die Säulenkombination gegeben. Anschließend wäscht man mit 1 000 ml des heißen Isopropanol-Wasser-Gemisches (5.3.3) die Säulen nach.
Zur Gewinnung der nichtionischen Tenside werden Durchlauf und Waschalkohol vereint und — vorzugsweise im Rotationsverdampfer — bis zur Trockne eingedampft. Der Rückstand enthält die BiAS. Entionisiertes Wasser zugeben, bis ein bestimmtes Volumen erreicht ist, und den BiA